Predicting drug-gene interaction of all FDA approved drugs using multi-modal neural networks
Scientists have identified over 100 medications for which known genomic variants play an important enough role to inform the guidelines for prescribing medications (NHGRI). This is very powerful, and it’s becoming a common medical practice to check patients’ genomes for mutations before administering certain drugs, to predict compatibility of the treatment. However, 100 medications is a very limited set of medicines for which we can do this. I recently did a deep dive on foundational deep learning models in biology, and I believe that based on that limited set of genetic variants and drugs we can likely predict drug-gene interaction of all FDA approved drugs, and possibly medicines that are still in development. The outcome of this is not only being able to provide recommendations to patients on treatment options based on predictive AI. I think this could actually be a great way to get us one step closer to modeling the internal behavior of a cell.
A short list of 184 alleles and their interactions with roughly 100 medicines is known, verified experimentally, and listed on a database called PharmCAT. An example is the HLA-B*57:01 allele, which encodes for a mutation in the HLA-B57 protein. Due to this mutation, the HLA-B57 protein now not only binds the HIV antiviral drug Abacivir but also binds a peptide which the body perceives as foreign, causing a hypersensitivity reaction. By knowing this we can design better HIV treatment plans, which require a messy combination of different antivirals. Yet, some critical genes are not listed on PharmCAT. For example CYP2C8, involved in the metabolism of NSAIDs (Ibuprofen), which is known to have mutations. We actually have a large amount of information available on the involvement of genes in pathways relevant to metabolism or other drug relevant cellular behavior like that of membrane molecules. Similarly, structural information on these small molecule drugs is well documented. And information on the metabolism of certain drugs, and their interactions with critical pathways, is available.
We could use all of this information and foundational protein models to predict unknown interactions between gene variants and drugs. All of this information can be processed and turned into machine readable format. From a variant calling file (VCF), mutant alleles and their natural occurring gene variant are retrieved one-by-one. Small molecule drugs are converted to SMILES format. And pathways are vectorized. Next, feature extraction is performed. Alleles are modeled into protein format either using Uniprot or Alphafold3 if necessary, and information on structure. For the drugs, 3D structure is derived from e-Drug3D or generated using molGNN or ChemBERTa, and features like are. It’s possible more complex models like DIFFDOCK-L could be used here, but I think they might not be necessary. These multi-modal features serve as input for a multi-input model, a graph neural net which combines them in a unified latent space. The model can learn from the complex web of interactions between proteins, and small molecules from an attention mechanism. The output would be an interactions score (IS). The model can be pretrained with known gene interactions from PharmCAT, that also have confirmed protein confirmations, and a loss function containing the MSE(IS) and a binary cross entropy loss for classification of positive or negative gene-drug interactions.
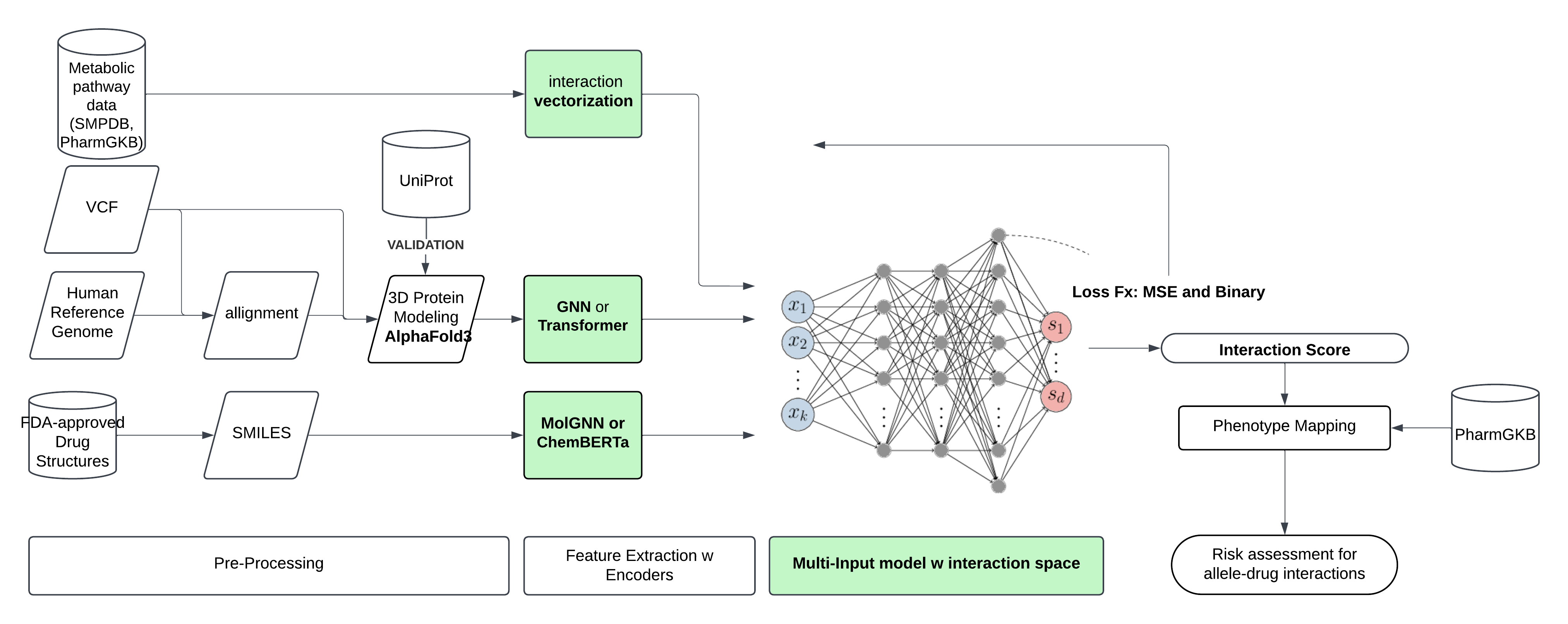
Once the model is trained, you could provide it with a set of unseen pharmacogenes that contain mutations compared to the reference human genome. The model would then predict medications that would be incompatible with these specific genomic variations. To wrap up this project, ideally we would want to test the model with a full human genome VCF file as input. I can also imagine that this model could somehow be fine-tuned by adding a new never seen before small molecule as input, making this a useful tool for drug development and clinical trial design.
I worked on this idea for about 2 days, during MIT 6.S191 Intro to Deep Learning. So do I know what I’m talking about? Not really, probably. But I see myself somewhere on the slope of enlightenment in the Dunning-Kruger effect. If I ever work out an MVP, you’ll be able to find it on my github.